https://preview.redd.it/1vvxii9ruw651.png?width=1303&format=png&auto=webp&s=c9ef7dc9d10f6d077e380449c6541a566406a70e
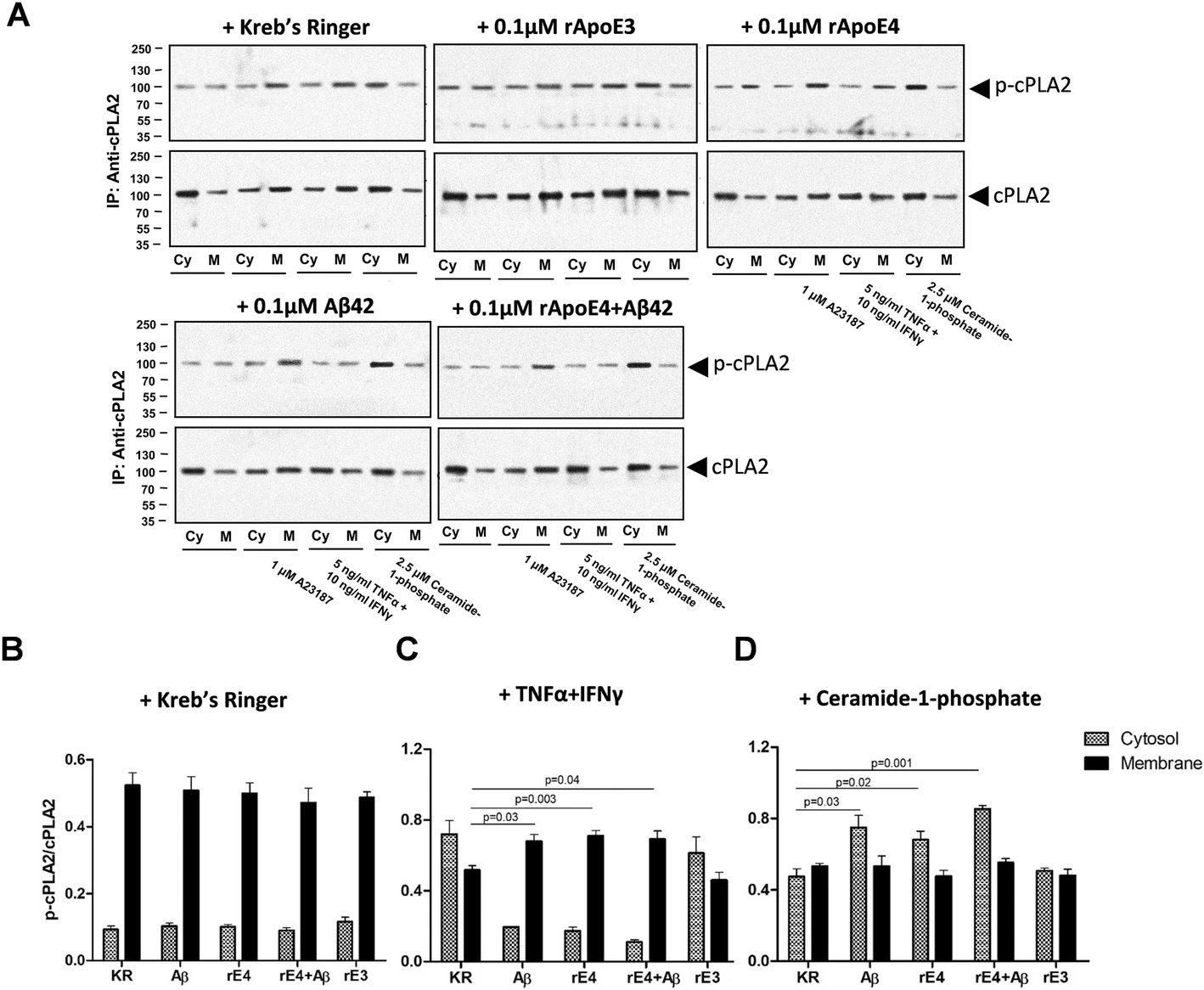
Let's say I have 5 wild type samples and 5 knockout samples to compare protein expression from. What is the best way to present this data on a western blot taking in to account biological and technical replicates? One way I have seen is pooling all the samples from each group together and running it in triplicate. Another way is running each sample individually, but there would not be any room on the gel to run technical replicates of each biological replicate.
Additionally, which way do journals prefer this? Most of the papers I have read are not clear on this in their western blot figures.
Thanks!

Hi there homeworkhelp, bit of a stickle here.
I'm currently in the process of quantifying the bands on my Western Blot membranes, in which the secondary antibodies were detected via fluorescence. The antibody band (polyclonal, Cav2.2) I'm trying to quantify has produced two bands at approximately 130 and 200 kDa - which is what it should do, as the manufacturer's example shows the same bands produced. I believe this may be because of the polyclonal antibody targeting different isoforms of the target subunit - but the manufacturer's website doesn't clarify why two bands are produced so I can only speculate (seems like a significant molecular weight difference?)
However, how do I quantify this? Combine the two bands' fluorescence values, or take an average of the two?
Or should I avoid quantifying at all; my project is focused on detecting the presence/abundance of Sodium channels, and this is a Calcium channel antibody I'm having a problem with. Perhaps I should not try to quantify this at all as it's not the main objective?


This is something that just absolutely blows my mind, and I cannot understand it yet. How were we able to develop Western Blots? The procedure seems so well put together, but I cannot understand the logic behind it. How did researchers go about figuring out a way to detect specific proteins using Western Blot?



This is a response to a person's statement on Twitter disparaging a non-simifulam western blot. Get ready, this response is a play in 3 acts. Protip: You can make your browser full screen, then click on an image below to get the hi-rez version, to review the compression swirls yourself.
Act 1: Here is the actual proper, original, full size image file direct from the source journal article, not starting from a jpeg with more artifacts added. (Thanks Derm95).
Act 2: Zoom in, enhance, as they say on the old CSI shows. This is a screenshot of just the tools that the Twitter person said she is using. I have better photo editing software than Preview (which is the Mac equivalent of Windows Paint), but the goal is for reproducibility in any bit of science. Here is a screenshot, featuring the settings to get there. This is all the blots, not just one.
Settings tweaks, with the data settings to reproduce (Click to see full size)
Act 3: And behold! There are compression swirls every which way, through bands, random locations, extending out from the smears of the bands. I am underwhelmed. When I look at the clouds, my favorites to pick out are a happy starfish and what seems to be a cat with a very long tongue, marked below.
My impression: This image has a lot of compression artifacts, and I do not feel very swayed towards any wrongdoing. If it makes some people feel better to rerun the gel and upload a raw image file, so be it. Someday, journals will likely be online only, with giant clinical x-ray sized images anyways, making compression artifact arguments moot in the future.
And instead of spending time on this, I w
... keep reading on reddit ➡
Titel.



Hi all. Just wondering if there is anyone in the New Orleans are that has had luck with finding a lab that will draw and prep your blood for Western Blot. I started calling on Friday and no luck yet.
UPDATE: I was able to get in touch with CPL main office in my area who said that I could just bring the kit in and the workers at whichever lab I go to would follow the instructions in the kit.


As in made all the arrangements, found a doctor to sign off on it, found a lab willing to cooperate? What was the experience?
I apologize if this is obvious. I’ve been struggling with an invisible illness for a long time that has the features of chronic fatigue or Lyme (extreme neck stiffness, joint pain, fatigue, migraines, brain fog etc). I had 1 single test “Lyme western blot” and never pursued it again. Now I realize these tests aren’t always reliable and there are other tests that can be done? Did anyone find they had this even after a negative like that? Thank you for the support!

wouldn't the SDS/boiling get rid of any proteases?
We are wanting to do something for Hel and we heard that a blot on halloween while wearing a mask works well.



Can Nupage gels be transferred with Tris-Glycine transfer buffer?
Also, it says the maximum loading volume for 12 well gel is 20ul. Is this a hard maximum? I would like to use 40ul.

I finally received my western blot results back yesterday and tested negative for both HSV1 and HSV2. I wanted to say thank you to all the individuals that contribute to the resources and knowledge in this community.
I was going through a routine OBGYN appointment and my doctor also recommended an STD test. I had no symptoms of any STDs and didn’t expect the test to come back with any positive results. On December 10, 2020 I tested positive for HSV2 (Igg level 1.58). I was negative for HSV1. I was shocked and pretty upset with myself. The test was performed by Quest diagnostics. I never had any symptoms but accepted the positive result and tried to move on.
Over the next several months, I read more online about the diagnosis, symptoms, and the possibility of a false positive. I got tested again on June 25, 2021 and tested positive for HSV2 again with Igg level 2.94. I thought for sure I have it and I’m just an asymptotic carrier. Since my Igg level had increased, it seemed a sure thing that I was an asymptomatic carrier. Again, I accepted it and continued with my life as normal.
I disclosed successfully to two partners since my initial diagnosis. The first partner did not think much of it at all and we had a very nice 10 month relationship. He never contracted any form of HSV. After recently (successfully) disclosing to a new partner, I decided that I would pursue the western blot test. It was a pain in the butt and I’m honestly shocked that my results are negative. I don’t really even believe it but I’m grateful this sub encourages everyone to go through with the test.
Anyway, if you test on the low end of positive via Quest Diagnostics and have no symptoms, definitely do the western blot test. Having a for sure result is the best thing! Even if it was a positive result, I would be happy to be confident in my status and know I’ve taken every step to inform myself and potential partners.
TLDR: 12/10/20 positive HSV2 Igg 1.58 6/25/21 positive HSV2 Igg 2.94 1/17/22 negative HSV2 by Western Blot

I’m running a Western Blot, and today I was supposed to develop the membrane but our imaging system is in need of repair (which I didn’t find out until after I developed the membrane and walked over to see that it wasn’t working).
The membrane has already been developed once; yesterday the imaging system was functional and after I got my images, I stripped the membrane and reprobed it overnight with the primary antibody. So today, I had washed and treated the membrane with the secondary.
The imaging system will undergo maintenance Monday morning, so until then, I’m thinking about washing the membrane with TBST, and then storing it in 4 C with TBS.
Come Monday, will I have to reprobe with the primary and secondary antibodies again or will I be able to simply develop the membrane again? I’m not sure if leaving it over the weekend in TBS will weaken signals. Any advice is appreciated! Thanks in advance!
Purifying a new N-His6 tagged protein in the lab, and I've run a decent-ish Western blot with all of my fractions (soluble, insoluble, etc.) to check for solubility, concentration efficiency, etc. that comes with finding out where a protein ends up.
My Western (blocked with BSA, blotted with anti-His6, stained with CN/DAB) shows that my protein (red arrow) exists, but also I have some pretty noticeable bands at lower MWs. I am fairly confident that the three most prominent lower bands on the leftmost lane are proteolysis products that retain the His-tag. The faint higher MW bands I am also fairly confident are non-specific staining.
Are the fainter lower MW bands background bands due to overstaining and/or non-specific staining? How can I get rid of those in the future? I was thinking decreasing antibody concentration, better and more thorough washes before staining, and maybe a better blocking step? Any help would be appreciated!
)
{kind=link}
{kind=link}
{kind=link}
{kind=link}